
For anyone working on an Abbreviated New Drug Application (ANDA), this isn't just a 'nice to have' anymore. The FDA basically made it a regulatory expectation back in 2011. While it takes more effort upfront, the payoff is huge. We're talking about a 23% increase in approval rates and review cycles that are nearly five months shorter. It transforms generic development from a 'copycat' game into a precise science.
The Core Components of a QbD Strategy
You can't just say you're doing QbD; you have to prove it through a specific technical architecture. It starts with the Quality Target Product Profile (or QTPP), which is essentially the 'blueprint' of the drug. If you're making a generic, your QTPP must show that your version is at least 95% similar to the Reference Listed Drug (the original brand-name version) in terms of how it performs in the body.
Once you have the blueprint, you identify Critical Quality Attributes (CQAs). These are the physical or chemical properties that must be right for the drug to be safe and effective. In a typical generic, you'll track 5 to 12 CQAs. A big one is the dissolution rate-the speed at which the drug dissolves-which needs a similarity factor (f2) greater than 50 compared to the original. If the drug doesn't dissolve right, it won't hit the bloodstream correctly, and you'll fail your bioequivalence tests.
Next come the Critical Process Parameters (CPPs). These are the 'knobs' you can turn during manufacturing. For example, how much moisture is in the granulation? If it's between 1.5% and 3.0%, you're good. If it hits 4%, the tablet might crumble. By using Design of Experiments (DoE), scientists can test multiple variables at once to see how they interact, rather than changing one thing at a time and guessing the result.
| Feature | Traditional Approach | QbD Approach |
|---|---|---|
| Process Logic | Fixed 'Recipe' (Single point) | Scientifically Justified Ranges |
| Quality Check | End-product testing | Real-time monitoring (PAT) |
| Regulatory Risk | Higher chance of CRLs | 31% fewer CRLs (FDA data) |
| Flexibility | Any change requires new filing | Design Space allows adjustments |
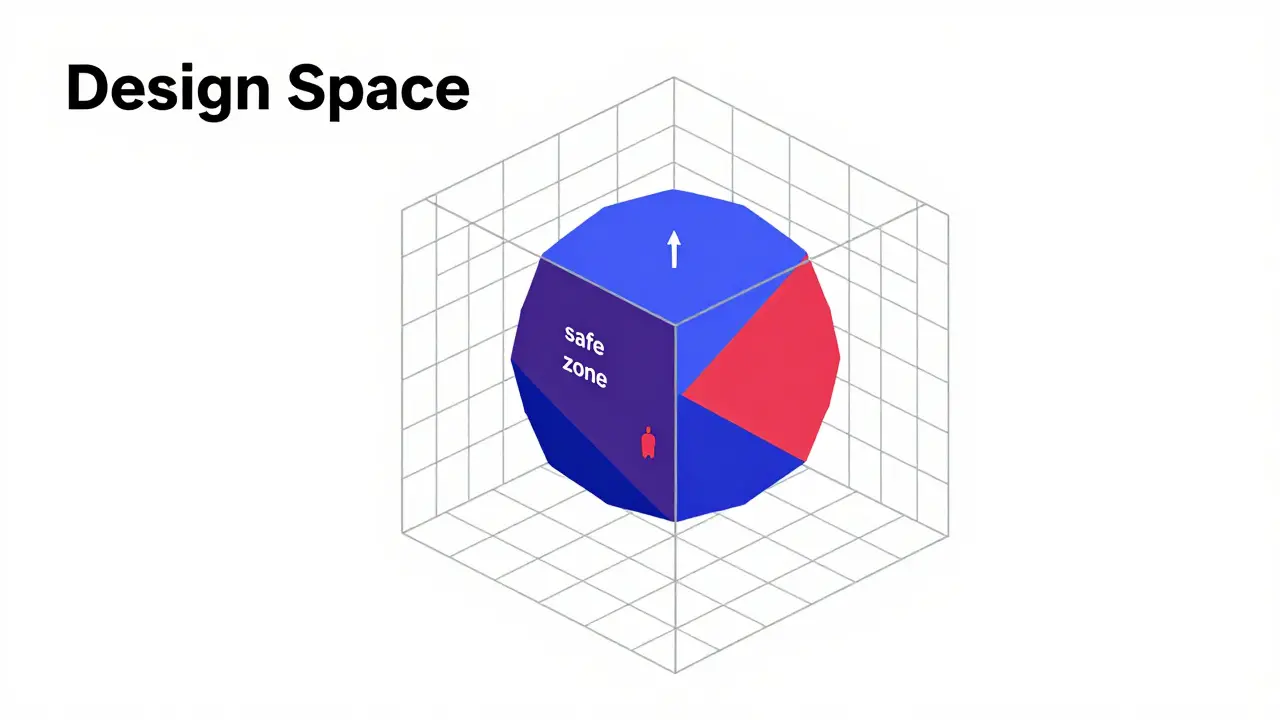
The Magic of the Design Space
The most powerful part of this whole system is the Design Space. Think of it as a 'safe zone.' If you can prove to the FDA that as long as your temperature is between 40-50°C and your pressure is 10-15 kN, the pill will always be perfect, then you've defined your design space. The amazing part? If you stay within that zone, you can change your settings without asking the FDA for permission every single time. This can save a company between $1.2 and $2.8 million a year in regulatory paperwork.
To keep this zone secure, companies use Process Analytical Technology (PAT). Instead of taking a sample to a lab and waiting two days for a result, PAT tools like near-infrared spectroscopy let you monitor the chemistry in real-time. It's like having a digital dashboard in a car that tells you exactly when the engine is overheating, rather than waiting for the car to smoke before you pull over.
Is QbD Always the Right Move?
Here is the reality: QbD isn't free. It's expensive and slow to set up. Initial development costs can jump by 25% to 40%, and you might add up to eight months to your timeline. If you're developing a very simple, low-cost immediate-release tablet that's already been made a thousand times, over-engineering it with QbD might be a waste of money. Some experts have pointed out cases where companies spent nearly half a million dollars on DoE studies for a product that didn't need that level of scrutiny.
However, if you're tackling "complex generics"-things like inhalers, transdermal patches, or modified-release tablets-QbD is almost mandatory. These products are notoriously finicky. If your in vitro-in vivo correlation (IVIVC) is off, your drug won't work in a human, no matter how good it looked in the lab. In these cases, the 28-42% increase in process robustness provided by QbD is the difference between a successful launch and a total failure.
Real-World Gains and Pitfalls
Let's look at how this actually plays out in the factory. One pharma professional noted that switching to a QbD-based strategy for a generic version of esomeprazole dropped their post-approval deviations from 14 per year down to just 2. That's not just a quality win; it's a financial win, saving nearly $850,000 in investigation costs alone. Another company used their approved design space to make 11 different manufacturing adjustments during the pandemic supply chain crisis without needing new regulatory approvals. This kept their delivery rate at 99.8% while others were facing stockouts.
But it's not all sunshine. The biggest hurdle is the talent gap. A scientist can't just 'wing it' with QbD; they need 80 to 120 hours of specialized training in Quality Risk Management and multivariate analysis software like MODDE Pro. Many firms struggle because they buy the expensive equipment but don't invest in the people, leading to a lack of "mechanistic understanding." If you don't actually understand why a certain temperature affects your drug, your design space is just a guess, and the EMA will catch that during review.
The Road to Implementation
If you're planning to move toward a science-based approach, don't try to do everything at once. Start by characterizing the Reference Listed Drug using advanced analytical tools-this can actually cut your development time by 30% by giving you a clearer target. If you're launching multiple strengths of the same drug, use a risk-based bracketing approach. Instead of doing full studies on every single dose, you study the extremes and the middle, which can cut your workload by 45%.
For the bold, moving toward continuous manufacturing is the next frontier. Instead of making drugs in batches, you have a constant flow. When combined with QbD, this can increase batch consistency by nearly 30%. We're seeing this already with products like levothyroxine, where the integration of design spaces and continuous flow has virtually eliminated batch-to-batch variation.
Does the FDA require QbD for all generic submissions?
While not strictly mandatory for every single simple drug, it is a strong regulatory expectation. For all ANDAs submitted after October 1, 2017, the FDA expects QbD elements to be present. For complex generics, it is virtually essential for approval.
What is the biggest financial risk of adopting QbD?
The primary risk is the high initial investment. Between specialized software, PAT tools (which can cost $500,000+), and extended development timelines (4-8 extra months), the upfront cost is significantly higher than traditional 'recipe-based' development.
How does a Design Space differ from a manufacturing specification?
A specification is a fixed limit (e.g., "Must be 40°C"). A design space is a multidimensional combination of variables (e.g., "Between 38°C and 42°C, as long as the pressure is 12kN"). Staying within the design space does not require a new regulatory filing, whereas changing a fixed specification does.
Can QbD help with bioequivalence failures?
Yes. By identifying Critical Quality Attributes (CQAs) and using DoE to optimize them, developers can ensure their product's dissolution and impurity profiles are nearly identical to the RLD, significantly reducing the risk of failing clinical bioequivalence trials.
What is the role of PAT in a QbD framework?
Process Analytical Technology (PAT) provides the real-time data needed to keep the process within the design space. Instead of testing a sample after the batch is finished, PAT allows for in-process adjustments, reducing end-product testing requirements by 35-60%.